The Wankowicz Lab investigates how entropy governs binding and catalysis using an integrated machine-learning, bioinformatics, and experimental framework.
Developing novel algorithms to detect conformational ensembles in experimental data
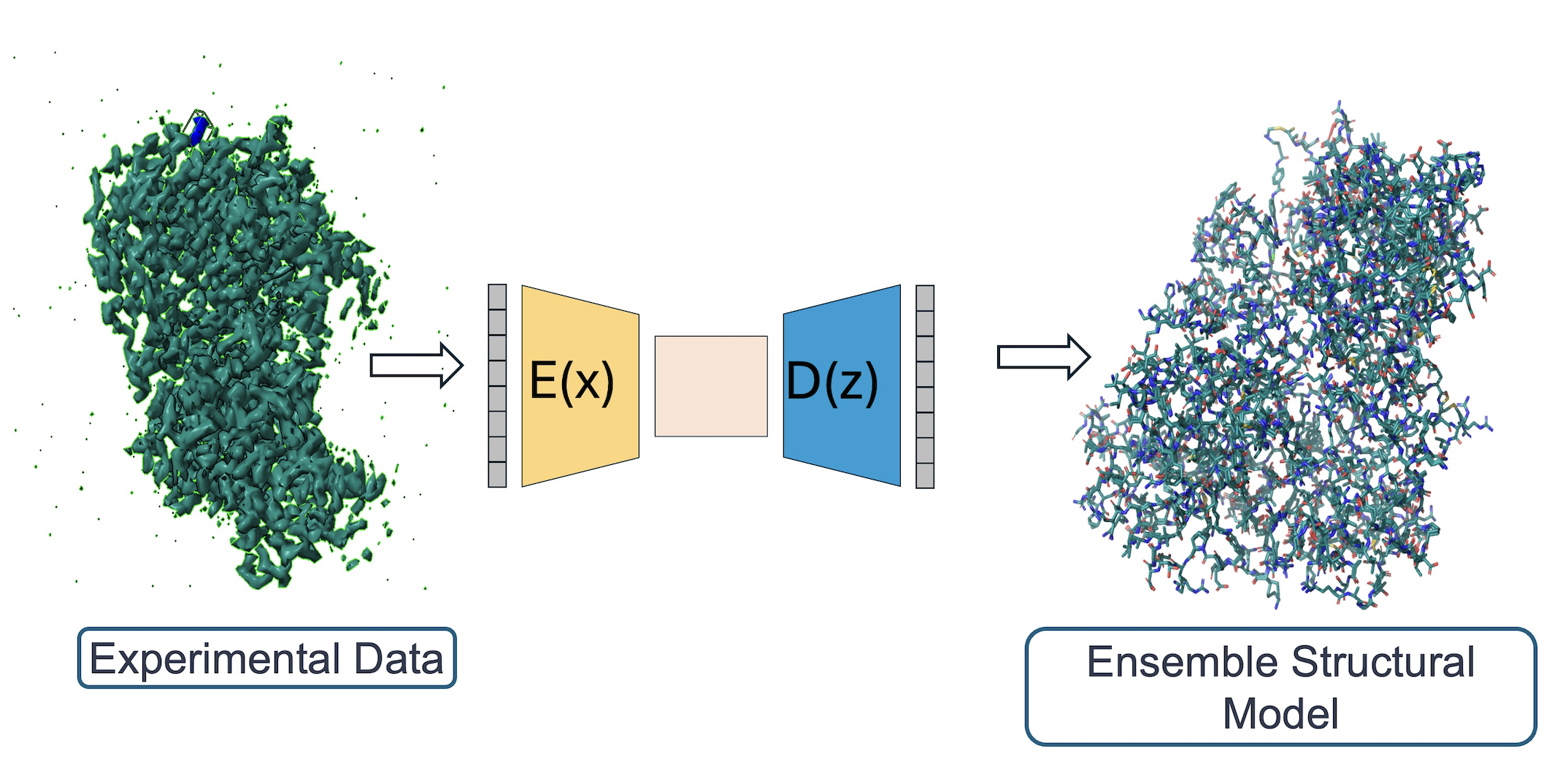
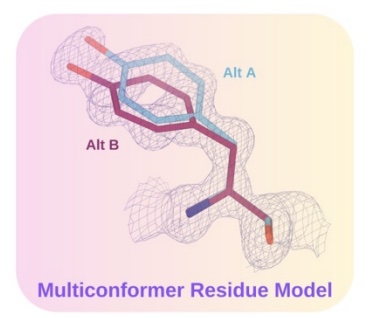
X-ray crystallography and single particle cryo-EM studies collect data averaged over tens of thousands to billions of individual system copies. Each molecule within the system can adopt a different conformation (conformational heterogeneity) and may differ slightly in chemical composition (compositional heterogeneity). However, structural models generally represent the system with a single coordinate set. This simplification overlooks the multiple states present in the experimental data and obscures entropy information.
We build algorithms to model the rich information hidden in experimental structural biology data, using machine learning and optimization. The lab develops a software package, qFit, which automatically detects more of the conformational ensemble and can improve the fit of the models to experimental data in high-resolution structures.
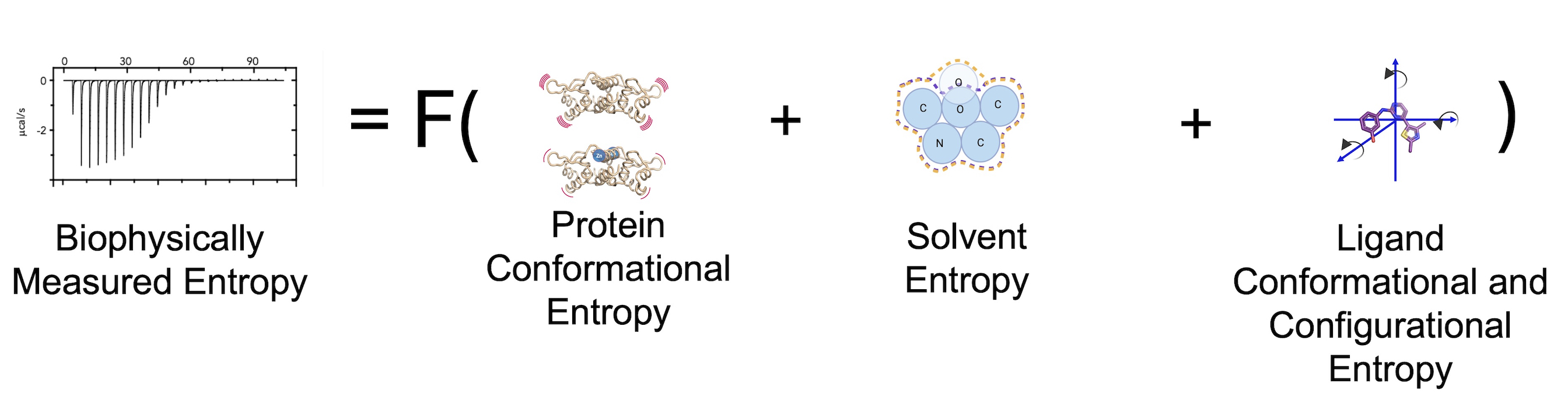
Connecting structures to thermodynamic biophysical binding measurements
Most information about protein function, binding affinity predictions, and mutations’ impact is based primarily on enthalpic parameters, encompassing only 50% of free energy. One key reason enthalpic changes are favored is that quantitative metrics can be extracted from structural models, enabling the easy use of these algorithms, such as binding prediction or ligand docking. However, equivalent metrics for entropy do not currently exist, preventing us from explicitly incorporating it into our structural intuition of binding. Our goal is to change this paradigm through computational, theoretical, and experimental approaches.
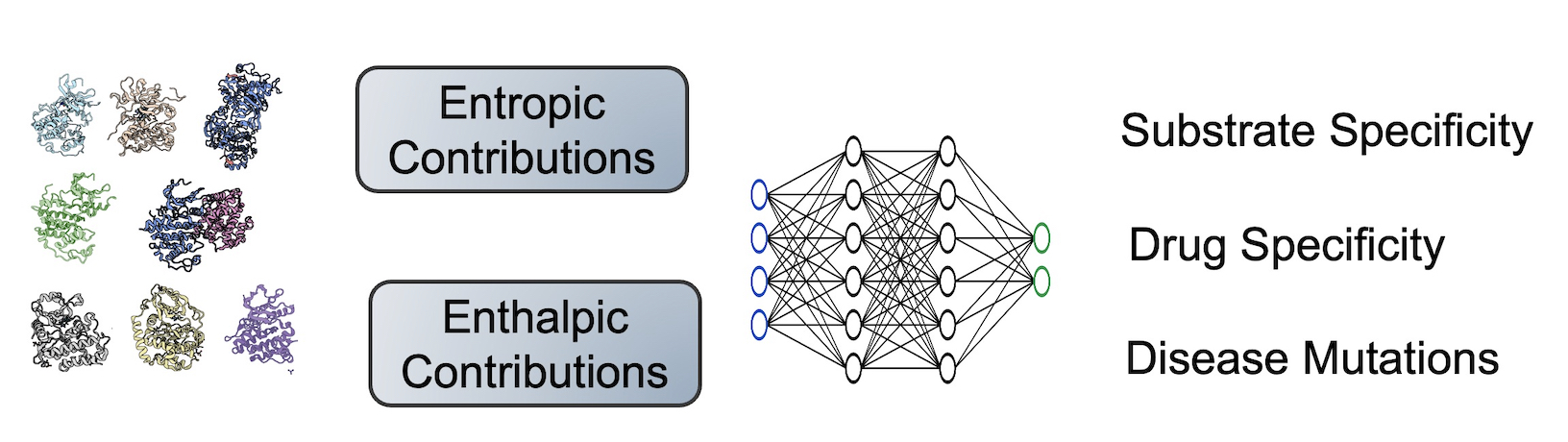
How does the entire conformational ensemble impact binding specificity and catalysis?
Binding specificity is foundational to the biochemistry of living organisms. However, our atomistic understanding of specificity is primarily driven by static interactions between a substrate and catalytic residues, while ignoring the critical role of protein dynamics. While functional residues almost always decrease their dynamics upon binding or during catalysis (reducing entropy), the rest of the protein scaffold can increase or decrease dynamics, leading to different entropic consequences for the entire enzyme.

Leveraging machine learning, bioinformatics, and experimental approaches, we aim to identify how conformational ensembles, encompassing enthalpic and entropic contributions, impact binding specificity and catalysis. Our overall goal is to elucidate the role of entropy in substrate specificity and catalysis, thereby providing a more comprehensive thermodynamic understanding of enzyme function.